| Hint | Food | 맛과향 | Diet | Health | 불량지식 | 자연과학 | My Book | 유튜브 | Frims | 원 료 | 제 품 | Update | Site |
|
내몸 ≫ 감각기관 ≫ 수용체 ≫ G수용체 GPCR, class C 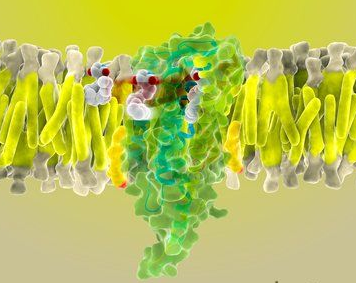 G protein 분류 : 역할 - Class A - Class B - Class C : 감칠맛 상승효과  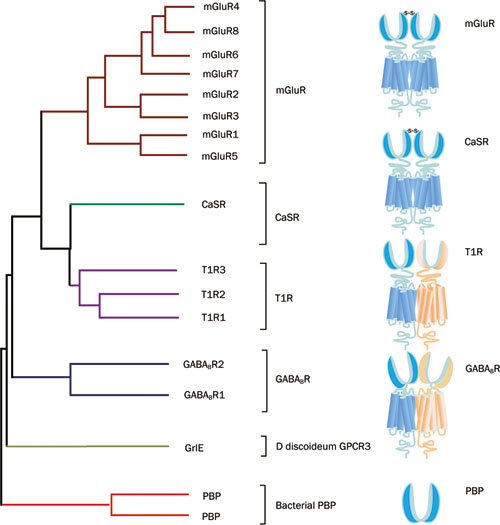 Class C GPCRs distinguish themselves from other GPCRs by two distinct structural features: first, they possess an unusually large extracellular domain that is responsible for orthosteric ligand recognition, while the 7TM (which normally contains the orthosteric ligand-binding site) has gained many allosteric sites; second, the functional class C GPCR molecules are obligatory dimers, so the interface between the VFT, CRD, and 7TM constitutes another important allosteric site. Furthermore, it was recently demonstrated that the VFT is large enough to accommodate allosteric modulatory sites adjacent to the orthosteric sites. The unique structure and complicated activation mechanism of class C GPCRs makes it possible to modulate their function by many new approaches. In recent years, allosteric modulation has become the most attractive approach because of the decreased side effects and development of patient tolerance, improved subtype selectivity and increased chemical accessibility. The development of allosteric modulators for class C GPCRs has progressed fast. Among them, Cinacalcet was the first clinical success. Following Cinacalcet, group I and II mGlu receptor modulators are expected to enter the market in the near future as the next generation of drugs that target class C GPCRs. By contrast, allosteric drugs that modulate the group III mGlu and GABAB receptors might represent a drug generation for the more distant future. To promote the application of allosteric modulation therapeutics that target class C GPCRs, future efforts should focus on investigating the precise structural dynamics and allosteric modulation mechanisms. Determination of the receptor structures is a direct way to address such issues. Traditional mutational analysis and chimeric constructs are also powerful tools that report on related information in the absence of a crystal structure for a particular receptor. Advanced functional assays, such as BRET (bioluminescence resonance energy transfer) or FRET (fluorescence resonance energy transfer), are widely used to reveal conformational changes and dimer or oligomer formation. Additionally, computational approaches, such as ligand- or structure-based homology modeling and docking, are gaining importance as valuable complements to experimental structure-function studies. These techniques, in combination with modern drug screening assays, make it possible to identify molecules targeting class C GPCRs through sites and mechanisms other then traditional orthosteric small molecules. L-Glutamate serves as the neurotransmitter at the majority of excitatory synapses in the mammalian CNS. As the metabotropic receptors for glutamate, mGlu receptors participate in the modulation of synaptic transmission and neuronal excitability throughout the CNS8, 9. The mGlu receptors are sub-classified into three groups based on sequence homology, G-protein coupling, and ligand selectivity9. Group I (mGlu1 and 5) couple to Gq/G11 and activate phospholipase Cβ, resulting in the hydrolysis of phosphoinositides and the generation of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol, whereas Group II (mGlu 2 and 3) and Group III (mGlu 4, 6, 7, and 8) couple predominantly to Gi/o, which inhibits adenylyl cyclase and directly regulates ion channels and other downstream signaling partners via the liberation of Gβγ subunits10. The widespread expression of mGlu receptors makes these receptors particularly attractive drug targets, and recent studies continue to validate the therapeutic utility of mGlu receptor ligands in neurological and psychiatric disorders, such as Parkinson's disease11, Fragile X syndrome12, Alzheimer's disease13, anxiety, and schizophrenia14. GABA is a major inhibitory neurotransmitter in the mammalian CNS. As the metabotropic receptor for GABA, GABAB receptor mediates slow and prolonged synaptic inhibition15. The GABABreceptor functions as an obligate heterodimer of two subtypes, GABAB1 and GABAB216, 17. GABAB1 contains the GABA binding site18, while GABAB2 is responsible for Gi/o protein activation19. In addition to a role in neuronal excitability and plasticity, GABAB receptor may promote neuron survival under conditions of metabolic stress20, ischemia21, or apoptosis22. This receptor is a promising target for the treatment of many diseases, including spasticity, neuropathic pain23, drug addiction, schizophrenia, anxiety, depression and epilepsy24, 25. The CaS receptor is a unique class C GPCR that can be activated by ions without the cooperation of other ligands4. This receptor is highly sensitive to a very slight change in extracellular Ca2+ concentrations, which ensures its significant role in regulating calcium homeostasis26. The CaS receptor is involved in several disorders, including hyperparathyroidism, osteoporosis and different forms of hypocalcemia26, 27, 28. The clinical success of Cinacalcet indicates that more efforts should be devoted to the discovery of novel ligands that modulate CaS receptor activation. The class C GPCRs contain three taste receptor subunits (T1R1, T1R2, and T1R3) that form two heterodimers, the sweet receptor (T1R2/T1R3) and the umami receptor (T1R1/T1R3)29, 30. Only cis activation occurs within the sweet and umami taste receptors, which means T1R2 in the sweet receptor or T1R1 in the umami receptor are involved in both orthosteric ligand recognition and in G protein activation, whereas the common subunit T1R3 loses the corresponding function31. In addition to natural sugars, the sweet taste receptor is also sensitive to artificial sweeteners, sweet proteins and some D-amino acids. In most mammals, the umami receptor can be activated by L-amino acids, whereas the human orthologue is only sensitive to monosodium glutamate and L-aspartate. Flavor enhancers, such as purine nucleotides, have the ability to potentiate umami receptor function. These artificial sweeteners and flavor enhancers represent a large food sector market6. 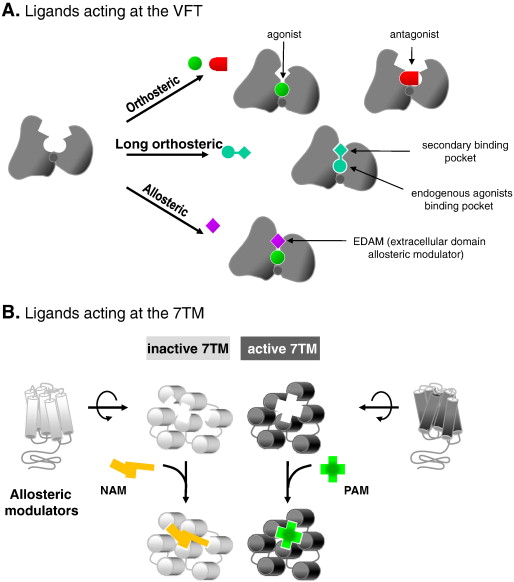  결합부위 Class C GPCRs contain multiple ligand interaction sites as a result of their particularly complicated structure and activation mechanism. These ligand binding sites are divided into two groups: orthosteric and allosteric binding sites. The endogenous ligand binding sites, or orthosteric sites, reside in the VFT domain. Both competitive agonists and antagonists interact with this site and induce significant conformational changes in the VFT: binding of a full agonist stabilizes a closed conformation35, whereas binding of competitive antagonists stabilizes an open conformation33, 34, 43. Binding of partial agonists results in a partial or a complete, yet unstable, closure of the VFT domain35, 58. In contrast, the allosteric sites are topographically distinct from the orthosteric sites in any given receptor. The binding of allosteric modulators changes the receptor conformation and, thereby, the affinities and/or efficacies of orthosteric ligands. In general, the positive allosteric modulators (PAMs) facilitate the action of the orthosteric agonists, whereas the negative allosteric modulators (NAMs) block the activation of orthosteric agonists by stabilizing the 7TM in an inactive conformation. The large extracellular domain and constitutive dimerization of class C GPCRs provide more potential allosteric sites compared with other GPCRs. To date, there are three groups of allosteric sites in class C GPCRs that have been reported (Figure 3).  Taste receptors — multiplicity of various ligand-binding sites A unique characteristic of taste receptors is their diversity of ligand-binding sites. Aside from the orthosteric sites, there are at least eight allosteric sites that have been identified in taste receptors: the EDAM sites for IMP in T1R1-VFT31 and for SE-2/SE-3 in T1R2-VFT67; the allosteric agonist sites for sweet proteins in T1R3-CRD45, cyclamate in sweet receptor T1R3 7TM70, S807 in T1R1 7TM31 and S819 in T1R2-7TM31; the PAM site for cyclamate in the umami receptor T1R3-7TM70; and the NAM site for lactisole in both the sweet and umami receptor T1R3-7TM71. These sites might represent potential targets for health-related products or drugs to treat diseases, such as hypertension or diabetes. 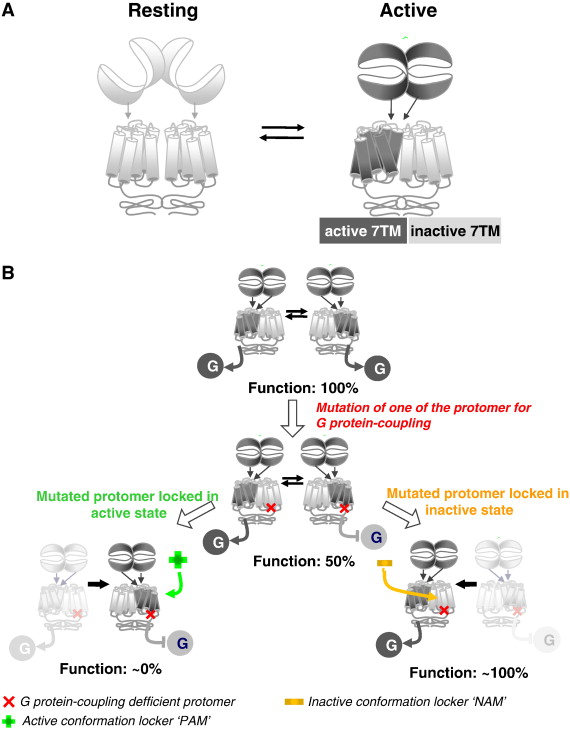 |
||||
|

|
|||